روز بیماری های خاص و صعب العلاج
براساس تعریف وزارت بهداشت، چهار بیماری تالاسمی، هموفیلی، دیالیز و ام اس در زمره بیماران خاص قرار میگیرند و درمان این بیماریها به عنوان درمانهای صعب العلاج به حساب می آید و عوارض بیماری تا پایان عمر همراه بیمار میماند.
بیماریهای سرطان، ام اس، پیوند کلیه، دیابت، اوتیسم، ای بی را نیز جزو بیماریهای صعب العلاج دستهبندی میکنند.
علت نامگذاری روز بیماریهای خاص
بنیاد امور بیماریهاى خاص به عنوان یک نهاد مردمى و غیردولتى، فعالیت خود را از ۱۸ اردیبهشت سال ۱۳۷۵ به منظور ساماندهى و ارتقاى وضعیت بیماریهاى خاص درزمینههاى مختلف درمانى، دارویى، آموزشى، پیشگیرى و اجتماعى آغاز کرد. از آنجا که متولى امر درمان درکشور، وزارت بهداشت درمان و آموزش پزشکى است و سیاستهاى بهداشتى و درمانى توسط این وزارتخانه تدوین میشود، بنیاد امور بیماری هاى خاص به عنوان یک سازمان غیردولتى و با بهره گیرى از کمکهاى مردمى برای بهبود وضعیت بیماری هاى خاص تلاش مىکند.
تلاشِ بنیاد امور بیماریهای خاص بر این بوده تا تمام مراحل درمانِ این بیماران، اولا درکشورِ خودمان و ثانیا به شکلِ کاملا رایگان انجام می شود.
بودجه و منابع مالی بنیاد بیماریهای خاص از کجا تامین می شود؟
بودجه و منابع مورد نیاز فعالیتها و برنامه هاى این بنیاد، به طور عمده ازطریق کمک افراد حقیقى و حقوقى و نهادهاى دولتى و غیردولتى تامین می شود.
بیماران خاص از نگاه آمار و ارقام
بر اساس آمارهای رسمی، حدود ۱۳۰ هزار بیمار خاص درکشور داریم که این تعداد صرفاَ شامل بیماران خاصی است که دولت آنها را شناسایی کرده است؛ در حالی که برخی بیماران خاص در کشور تحت پوشش و حمایت هیچ نهاد دولتی قرار ندارند؛ به همین دلیل آمار دقیقی از تعداد بیماران شناسایی نشده؛ در دسترس نیست.
بیماران خاص چه مسائل و مشکلاتی دارند؟
با آن که دولت و مراکز خیریه غیردولتی برای تامین دارو و ادامه فرآیند درمان به این بیماران کمک میکنند، اما این کمک ها در حدی نیست که جوابگوی همه نیازهای واقعی بیماران خاص باشد و مجموع مشکلات آنها موجب شده بسیاری از این بیماران نتوانند بدون مشکل مالی به ادامه روند درمان فکر کنند.
ـ آزمایشهای منظم و دورهای و هزینههای سنگین آن.
ـ کمبود داروی بیماران خاص و صعب العلاج در داروخانه و افزایش سرسام آور قیمت دارو.
ـ فراهم نبودن فضای شغلی که این گروه از بیماران بتوانند قابلیت های شغلی خود را بروز دهند.
ـ تاکید برتسهیل بازنشستگی زودهنگام بیماران خاص.
ـ هزینههای درمانی بسیار زیاد و هزینههای عوارض بیماری که هزینههای دیگر را نیز ایجاد میکند .
ـ طرد شدن بیماران مبتلا به ام اس از کانون خانواده و طلاق آنها (زنان) توسط همسرانشان معضل دیگری است که بارمشکلات ذهنی و روحی آنان را بیشتر میکند.
ـ از دست دادن شغل پس از ابتلا فرد به بیماری.
برای پیشگیری از ازدیاد بیماران خاص و نیز برای کمک به این بیماران چه کارهایی باید انجام داد؟
پرهیز از ازدواجهای فامیلی
توجه به آزمایشات قبل از ازدواج و بارداری
طرحِ غربالگریِ به موقع نوزادان در تمام نقاط کشور
راهاندازیِ ایستگاههای چکاپ سرپایی در مدارس و دانشگاهها و خیابان و ایستگاههای مترو در تمام نقاط کشور
بیمه رایگانِ بیماریهای خاص شامل تمامیِ موارد درمانی و دارویی این بیماران در سراسر کشور
افزایش تعداد کلینیک های درمانیِ بیماریهای خاص
افزایش تعداد واحدهای مددکاری در سراسر کشور
تحتِ پوشش قرار دادن کامل تمام بیماران خاص در سراسر کشور
تالاسمی
تالاسِمی (نام دیگر آن کم خونی مدیترانه ای است ) از بیماریهای خاص و یکی از بیماریهای ژنتیکی و یک اختلال خونی ارثی بوده که در آن، تعداد هموگلوبینها و گلبولهای قرمز در بدن فرد مبتلا به این بیماری از حالت نرمال کمتر است. هموگلوبین پروتئینی در گلبولهای قرمز است که آنها را قادر به حمل و انتقال اکسیژن میسازد. شمار پایین هموگلوبین و سلولهای خونی، میتواند موجب کمخونی شود.
هموگلوبین بخش انتقالدهنده اکسیژن در گلبولهای قرمز خونی است. هموگلوبین شامل دو زیرواحد مختلف به نامهای آلفا و بتا است.
اگر بدن شخص توانایی تولید کافی از هر نوع زیرواحد را نداشته باشد، سلولهای خونی بهطور کامل شکل نمیگیرند و توانایی انتقال اکسیژن بدن کاهش مییابد. در نتیجه نوعی کمخونی ایجاد میشود که تالاسمی نام دارد. هر چند تالاسمی یک اختلال منفرد نیست، اما یک گروه اختلالات از طرق مشابه بدن انسان را درگیر میکند. درک تفاوت میان گونههای مختلف تالاسمی مهم و ضروری است.
تالاسمی آلفا
افرادی که در آنها به میزان کافی زیرواحد پروتئینی آلفا تولید نمیشود به تالاسمی آلفا مبتلا میشوند. تالاسمی آلفا بهطور شایع در آفریقا، خاورمیانه، هند، آسیای جنوبی، جنوب چین و نواحی مدیترانه یافت میشود.
۴ گونه تالاسمی آلفا وجود دارد که با توجه به آثار آنها بر بدن از خفیف تا شدید تقسیمبندی میشود.
مرحله حامل خاموش
در این مرحله عموماً فرد سالم است، زیرا کمبود بسیار کم زیرواحد پروتئینی آلفا بر عملکرد هموگلوبین تأثیر نمیگذارد. به علت تشخیص مشکل، این مرحله حامل خاموش نامیده میشود. هنگامیکه فرد به ظاهر طبیعی صاحب یک فرزند با هموگلوبین H یا صفت تالاسمی آلفا میشود، مرحله حامل خاموش تشخیص داده میشود.
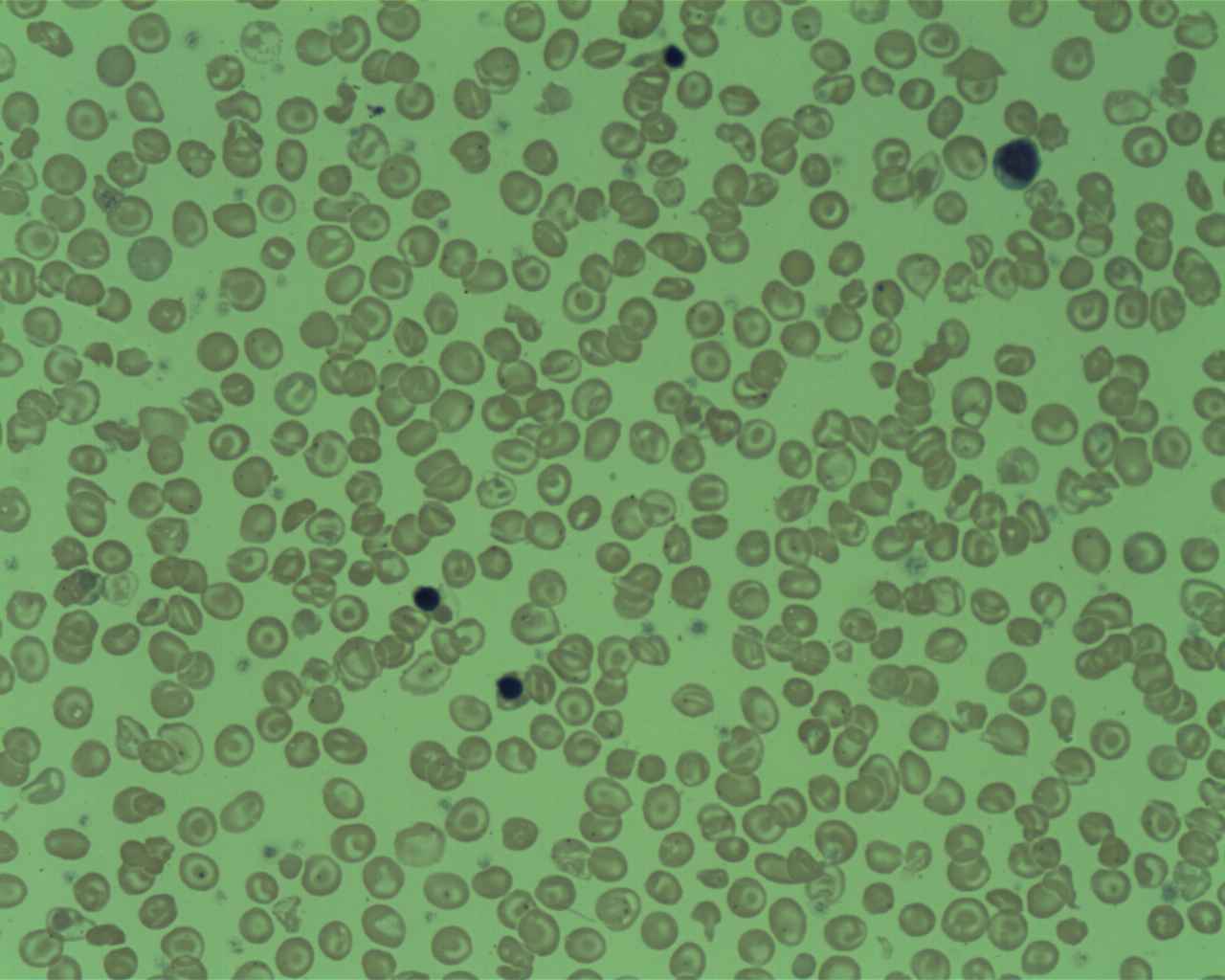
عکس خون محیطی از یک بیمار مبتلا به تالاسمی دلتا بتا
Hemoglobin Constant Spring هموگلوبین کنستانت اسپرینگ
یک فرم غیرمعمول از مرحله حامل خاموش که به واسطه جهش در زیرواحد آلفا رخ میدهد. علت این نامگذاری آن است که این موضوع در منطقهای در جامائیکا به نام کنستانت اسپرینگ کشف شدهاست. بیمار، همانند مرحله خاموش، هیچ گونه مشکلی را تجربه نمیکند.
صفت تالاسمی آلفا یا تالاسمی آلفا خفیف
در این مرحله کمبود زیرواحد آلفا بیشتر است. بیماران در این مرحله سلولهای قرمز خونی کمتر و کوچکتری دارند، اگر چه بسیار از بیماران علائمیاز بیماری را تجربه نمیکنند. پزشکان اغلب تالاسمی آلفا خفیف را با کم خونی فقر آهن اشتباه میگیرند و برای بیماران آهن تجویز میکنند. آهن هیچ تأثیری بر درمان کم خونی تالاسمی آلفا ندارد.
بیماری هموگلوبین H
در این مرحله، کمبود زیرواحد پروتئینی آلفا به حدی است که منجر به کم خونی شدید و بروز مشکلاتی نظیر طحال بزرگ، تغییرات استخوانی و خستگی میشود. نامگذاری به علت زنجیره H غیرطبیعی است که سلولهای قرمز خون را تخریب میکند.
هیدروپس جنینی یا تالاسمی آلفای ماژورتالاسمی آلفای بزرگ
در این حالت، در بررسی DNA فرد ژنهای آلفا مشاهده نمیشود. این اختلال باعث میشود گلوبین گامای تولیدی در جنین هموگلوبین غیرطبیعی بارت (Barts) ایجاد کند. بسیاری از این بیماران قبل یا در فاصله کوتاهی بعد از تولد میمیرند. در موارد بسیار نادری که بیماری قبل از تولد تشخیص داده میشود، تزریق خون داخل رحمی منجر به تولد کودکی با هیدروپس جنینی میشود. این نوزاد در سراسر زندگی خود به تزریق خون و مراقبتهای پزشکی نیازمند است.
تالاسمی بتا
در افرادی ایجاد میشود که در بدن آنها زیرواحد پروتئینی بتا کافی تولید نمیگردد. تا کنون دهها جهش در ژن HBB شناخته شدهاند که میتوانند موجب تالاسمی بتا گردند. این بیماری در مردم نواحی مدیترانه نظیر یونان و ایتالیا، ایران، شبه جزیره عربستان، آفریقا، جنوب آسیا و جنوب چین یافت میشود. مطالعات زیادی انجام گرفتهاست تا مشخص شود که در هر یک از این نواحی کدام جهشهای ژنتیکی در مبتلایان متداولتر است.سه گونه تالاسمی بتا وجود دارد که با توجه به آثار آنها بدن از خفیف تا شدید تقسیمبندی میشوند.
بیشتر بخوانید
🎥ویدئو/ مواد غذایی مفید و موثر در درمان کمخونی
🎙️سیف کست ۴-۴ – طرح سلامت، ایمنی و محیط زیست | امداد و کمک های اولیه – بخش چهارم
روشهای نوین مقابله با سرطان؛ از پیشرفت تازه در مهار لوسمی تا شخصیسازیِ درمان
تالاسمی مینور یا صفت تالاسمی
در این حالت کمبود پلی پپتید به حدی نیست که باعث اختلال در عملکرد هموگلوبین شود. یک فرد با این بیماری حامل صفت ژنتیکی تالاسمی است. این فرد به جز یککم خونی خفیف در برخی موارد، مشکل دیگری را تجربه نخواهد کرد. همانند تالاسمی آلفای خفیف، گاهی ممکن است پزشک بخواهد فرد مبتلا به تالاسمی بتای مینور را به عنوان علامتی از کم خونی فقر آهن با تجویز نادرست مکمل آهن درمان کند.
تالاسمی بینابینی
در این حالت کمبود زیرواحد پروتئینی بتا در هموگلوبین به اندازهای است که منجر به کم خونی نسبتاً شدید و اختلالات قابل ملاحظهای در سلامت فرد نظیر بدفرمیهای استخوانی و بزرگی طحال میشود. در این مرحله طیف وسیعی از علائم وجود دارد. تفاوت کم بین علائم تالاسمی بینابینی و فرم شدیدتر (تالاسمی ماژور) یا تالاسمی بزرگ میتواند گیجکننده باشد.
به دلیل وابستگی بیمار به تزریق خون، فرد را در گروه تالاسمی ماژور قرار میدهند. بیماران مبتلا به تالاسمی بینابینی برای بهبود کیفیت زندگی و نه برای نجات یافتن، به تزریق خون نیازمندند.
تالاسمی ماژور یا کم خونی Cooley’s Anemia
این مرحله شدیرترین فرم تالاسمی بتا میباشد که کمبود شدید زیرواحد پروتئینی بتا در هموگلوبین منجر به یککم خونی تهدیدکننده حیات میشود و فرد به انتقال خون منظم و مراقبتهای طبی فراوانی نیازمند میشود.
انتقال خون مکرر در طول عمر منجر به تجمع بیش از حد آهن میشود که باید توسط تجویز عوامل Chelator کمککننده در دفع جهت جلوگیری از مرگ و نارسایی ارگانها درمان شوند.
دیگر اشکال تالاسمی
به جز تالاسمی آلفا و بتا، اختلالات وابسته دیگری وجود دارند که به علت وجود ژنهای غیرطبیعی در کنار ژنهای آلفا و بتا یا جهش ژنی رخ میدهند.
- هموگلوبین E یکی از شایعترین هموگلوبینهای غیرطبیعی است و اغلب در مردم آسیای جنوب شرقی نظیر کامبوج و تایلند دیده میشود. هنگامیکه با تالاسمی بتا همراه شود بیماری بتا تالاسمی E ایجاد میشود که نوعی کم خونی نسبتاً شدید است که علائم مشابه با تالاسمی بتای بینابینی دارد.
- بتا تالاسمی داسی شکل
این حالت به علت وجود همزمان تالاسمی بتا و هموگلوبین S (هموگلوبین غیرطبیعی که در بیماران کم خونی داسی شکل دیده میشود). این بیماری بهطور شایع در نواحی مدیترانهای نظیر ایتالیا، یونان و ترکیه دیده میشود.
شدت بیماری به میزان تولید گلوبین بتا طبیعی توسط ژن بتا بستگی دارد.
هنگامیکه ژن بتا گلوبین بتا تولید نکند، شرایطی مشابه با کم خونی داسی شکل ایجاد میشود. هرچه گلوبین بتای بیشتری توسط ژن بتا تولید شود، شدت بیماری کاهش مییابد.
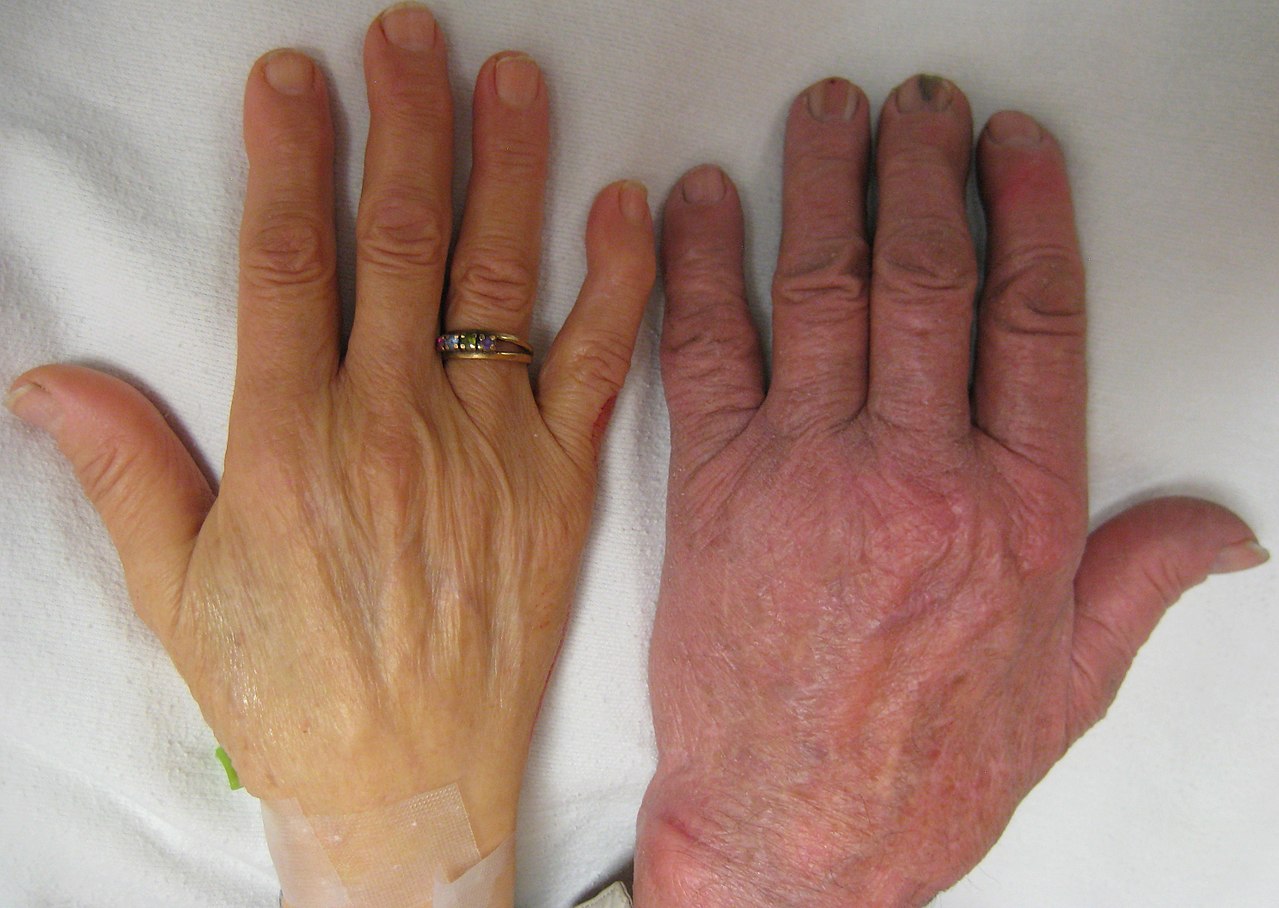
سمت چپ: دست فرد مبتلا به کم خونی شدید. سمت راست: دست فرد بدون کم خونی.
درمان تالاسمی
بیماران مبتلا به تالاسمی خفیف پس از تشخیص بیماری نیازی به مراقبتهای پزشکی و درمانی ندارند. افرادی که مبتلا به تالاسمی نوع بتا هستند باید بدانند که ممکن است بیماریشان با کمخونی ناشی از فقر آهن که بیماری شایعتری نسبت به تالاسمی است اشتباه گرفته شود. این دسته از بیماران باید از مصرف مکملهای آهن اجتناب کنند، اگرچه امکان وقوع کمبود آهن در آنها در هنگام بارداری یا خونریزیهای مزمن وجود دارد. مشاوره برای همه افرادی که دچار اختلالات ژنتیکی هستند توصیه میشود به خصوص زمانی که اعضای خانواده در معرض انواع شدید بیماری قرار دارند که قابل پیشگیری باشد.
کمخونی
بیماران مبتلا به تالاسمی شدید نیازمند مراقبتهای پزشکی هستند. جهت افزایش طول عمر بیماران انتقال خون به عنوان قدیمیترین اقدام مؤثر مطرح است.این تزریق خون برای فراهم آوردن مقادیری از سلولهای قرمز خونی سالم و هموگلوبین طبیعی که قادر به انتقال اکسیژن باشد، ضروری است.
در صورتی که بیمار تالاسمیک در گذشته به اندازه کافی خون دریافت نکرده باشد، لازم است جهت بهبود کیفیت زندگی وی، دفعات تزریق خون افزایش یابد. امروزه بسیاری از بیماران تالاسمی ماژور در هر دو یا سه هفته خون دریافت میکند، مقداری معادل ۵۲ پاینت خون در یک سال. Pint معادل واحد تزریق مایع تقریباً برابر ۴۷۳ میلیلیتر است.
افزایش بیش از حد آهن
از آنجاییکه راه طبیعی جهت حذف آهن در بدن وجود ندارد، چندین بار تزریق خون میتواند منجر به تجمع بیش از حد آهن در بدن شود و وضعیتی به نام «افزایش غیرطبیعی آهن» (Iron Overload) ایجاد کند. آهن مازاد برای بافتها و ارگانهای بدن به ویژه قلب و کبد سمی است و منجر به مرگ زودرس یا نارسایی ارگانها در فرد بیمار میشود. اضافه بار آهن به ویژه در قلب، مهمترین علت مرگ و میر بیماران تالاسمی ماژور میباشد.
برای کمک به دفع آهن اضافی، بیمار تحت درمان با دستهای از داروها به نام داروهای شلاتهکننده آهن (Iron-chelating agents) قرار میگیرد. این داروها از طریق فرایند چنگاله (به انگلیسی:Chelation شنیدن تلفظ) به آهن متصل میشوند و با دفع آهن متصل شده میزان آهن ذخیرهشده در بدن را با دفع آن از طریق ادرار یا مدفوع کاهش میدهند. زمان آغاز این درمان در بیماران تالاسمی بتا عامل بسیار مهمی در بقای آنان به حساب میآید و توانستهاست امید به زندگی در بیماران تالاسمی بتا را بهبود بخشد. مهمترین داروهای این دسته که مورد تأیید سازمان غذا و داروی آمریکا قرار گرفتهاند دفراسیروکس، دفریپرون و دفروکسامین هستند.دفریپرون (فریپروکس) به صورت قرص ۵۰۰ میلیگرمی و محلول خوراکی (۱۰۰ میلیگرم در ۰٫۴ میلیلیتر) وجود دارد. دوز رایج روزانه این دارو ۷۵ میلیگرم به ازای هر کیلوگرم و در سه دوز منقسم است. دفراسیروکس که نخستین داروی خوراکی مورد تأیید سازمان غذا و داروی آمریکا به منظور دفع آهن اضافی است به صورت قرصهای جوشان (اکسجید) ۱۲۵میلیگرمی، ۲۵۰ میلیگرمی و ۵۰۰ میلیگرمی که نیاز به حل کردن در آب دارند و قرصهای روکشدار (جیدنیو) ۹۰، ۱۸۰ و ۳۶۰ میلیگرمی وجود دارد. دوز پیشنهادی روزانه دفراسیروکس جوشان ۲۰ میلیگرم به ازای هر کیلوگرم تا نهایت ۴۰ میلیگرم به ازای هر کیلوگرم است که با معده خالی باید مصرف شود. دوز پیشنهادی روزانه دفراسیروکس روکشدار ۱۴ میلیگرم به ازای هر کیلوگرم تا ۲۸ میلیگرم به ازای هر کیلوگرم است که با یا بدون یک وعده غذای سبک میتواند مصرف شود.[۱۹] دفروکسامین (دسفرال) به شکل پودر لیوفیلیزه برای تزریق در دوزهای ۵۰۰ میلیگرمی و ۲ گرمی وجود دارد. این دارو به شکل زیرجلدی با دوز ۴۰–۲۰ میلیگرم به ازای هر کیلوگرم و در طی ۲۴–۸ ساعت و از طریق پمپ قابل حمل تزریق میشود. دوز مصرف وریدی دفروکسامین ۵۰–۴۰ میلیگرم به ازای هر کیلوگرم در روز و طی ۱۲–۸ ساعت برای ۵ روز هفته است. دوز مصرف عضلانی دفروکسامین ۱–۰٫۵ گرم روزانه است.

مشکلات تحمل درمان
تحمل درمان، برای بیماران مبتلا به تالاسمی یک امر حیاتی محسوب میشود. عدم تحمل این درمان منجر به مشکلات سلامتی شدید و مرگ زودرس در بیماران میشود. برای مقابله با مشکل عدم تحمل دارو، محققین به دنبال داروهای جدیدی هستند که بیمار آن را به آسانی تحمل نماید.
پیشگیری
در حال حاضر موثرترین راه پیشگیری از بیماری تالاسمی غربالگری این بیماری در سطح جمعیتی و سپس بررسی ملکولی افرادی میباشد که در غربالگری در زمره افراد تحت خطر طبقهبندی شدهاند. برای این منظور در ایران برنامه کشوری غربالگری تالاسمی توسط وزرات بهداشت در حال انجام میباشد.
مشاوره ژنتیک
مشاوره ژنتیک در افرادی که در غربالگری در زمره افراد تحت خطر طبقهبندی شدهاند یا افرادی که دارای بستگان نزدیک مبتلا به تالاسمی هستند توصیه میشود. ریسک تکرار این بیماری در فرزندان بعدی یک زوج دارای فرزند مبتلا ۲۵٪ میباشد که در صورت تعیین جهش در فرزند مبتلا و تأیید ناقل بودن والدین میتوان با تشخیص قبل از تولد در طی دوران بارداری از وضعیت ابتلا یا عدم ابتلای جنین اطمینان حاصل نمود.
انتهای پیام/